When running a cell relaxation, it is sometimes nice to monitor what is going on with the cell parameters. If you run the cell relaxation with Quantum Espresso, you can use my python script “pw2cellvec” to parse all the information you need.
So, first of all, run a “vc-relax” calcuculation with a large (about 2 times bigger) cutoff for both wavefunction (ecutwfc) and charge (ecutrho) and wait for a couple of iterations. These calculations are very expensive, due to the large cutoffs.
Then, just run the script and you will get something like this:
user> pw2cellvec.py -o pwout
- -
AA a= 6.02104498011 b= 6.02109150665 c= 9.45688483193 Vol.= 296.919819153
au a= 11.3781260524 b= 11.3782139748 c= 17.8709224124 c/a= 1.57063846279
alpha= 89.9989981833 beta= 90.0012313781 gamma= 119.99694776
- -
AA a= 6.02104759941 b= 6.02109604694 c= 9.45688817964 Vol.= 296.920644451
au a= 11.3781310022 b= 11.3782225547 c= 17.8709287386 c/a= 1.57063833552
alpha= 89.9989563444 beta= 90.0012827265 gamma= 119.996825039
- -
AA a= 6.02105023076 b= 6.02110063535 c= 9.45689158155 Vol.= 296.921479759
au a= 11.3781359747 b= 11.3782312256 c= 17.8709351673 c/a= 1.57063821412
alpha= 89.998913741 beta= 90.0013350646 gamma= 119.996700532
- -
AA a= 6.02105288621 b= 6.0211052793 c= 9.45689504969 Vol.= 296.922326733
au a= 11.3781409928 b= 11.3782400014 c= 17.8709417211 c/a= 1.57063809742
alpha= 89.9988702917 beta= 90.0013883718 gamma= 119.9965741346.02105288351 0.000164884388069 -7.29501276554e-05
-3.01038364832 5.21452769394 5.93607880947e-05
-0.000114579145698 4.15025775775e-05 9.4568950489
For each iteration, three lines are printed:
- 1st one, starting with “AA”, contains the lattice parameters in Angstroms (Å) and the cell volume in Å3
- 2nd one, starting with “au”, the cell parameters in atomic units (bohr) and the “c/a” ratio (useful for copy/paste to a new input)
- 3rd one, the alpha, beta and gamma (α, β and γ) angles in degrees (°)
After the last iteration in the output (even if the calculation is still running), the lattice vectors (in Å) are printed. Useful, i.e. when we need them for something else.
Finally, a tip on how to visually check the convergence in real time. With the following command, you can check what is going on very quickly with gnuplot:
user> echo "p '< pw2cellvec.py |grep au ' u 3 w l, '' u 5 w l" | gnuplot -persistThe previous command monitors cell parameter a and b for each iteration, but you can switch between columns 3, 5, and 7 for monitoring a, b and/or c.
The resulting plot for a and b looks like this:
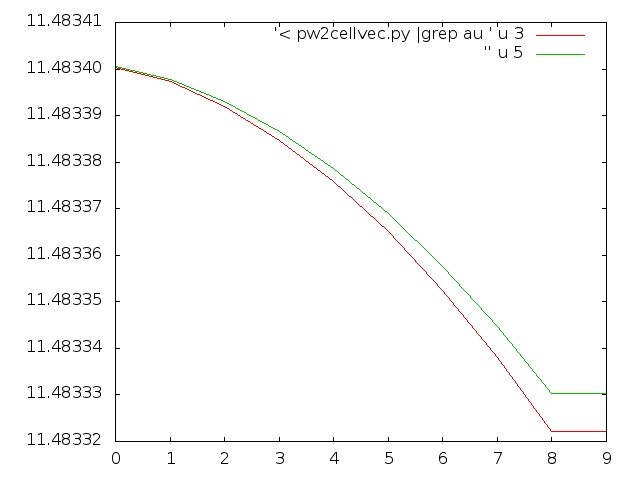
Graphical view of the convergence of the cell parameters over the relaxation iterations.
Dear Julen,
as a QuantumEspresso novice, I have found your website with additional resources and scripts very useful. However, I am wondering about your recommendation to use very large cutoffs in the vc-relax calculations. As far as I remember, I have never encountered recommendations to increase the cutoffs in such a drastical manner in the documentation of other plane-wave based DFT programs.
Could you please comment on the reasons for this recommendation? Furthermore, would you also recommend to run an optimisation with a fixed cell with a higher cutoff than a single-point calculation (assuming that it has been ensured that the single-point calculation is sufficiently converged with respect to the cutoff)?
Thank you and best wishes,
Michael
Dear Michael,
As you can see from the E vs cutoff graph in ““, we could run some rough and cheap relaxation even at 30 Ry. but to make sure we add 10 extra Ry to get a better value. If we would chose 50 Ry. the result would be slightly better, but the computational price to pay for this little improvement is usually not worth it, specially for a regular relaxation.
In the case of a cell relaxation if the forces are not accurate, the stress tensor won’t be either, and it the calculation can keep trying new atomic positions and cell vectors for really long, making it much more expensive than just increasing the cutoffs.
I said 3 times, but well… probably increasing the cutoffs to the double, should be enough too. If you want to make sure you use an optimum value, you can compare the stress tensor (pressure) at different cutoffs and whenever it shows to be really converged, that’s it.
Hmm… I should include an option in the “pw_cutoff” script to check for the convergence of the stress tensor…
Best regards
Julen
Dear Julen,
Thank you for sharing your experience. I am not familiar with python. I tried to run your script but I got an error:
Traceback (most recent call last):
File “pw2cellvec.py”, line 53, in
alat=float(line.split()[2].split(“)”)[0])
IndexError: list index out of range
I appreciate if you could help me. Thanks
Dear Farnoosh,
The quantum espresso output should output your cell parameter as “CELL_PARAMETERS (units= NUNMBER)”
Try this:
grep -a CELL_PARAMETERS out |tail -1With one of my output files I get this result for the previous command:
CELL_PARAMETERS (alat= 11.36100000)The error message raises because the script tries to parse the “alat” variable but your line containing the key “CELL_PARAMETERS” doesn’t probably contain anything else afterwards.
Are you defining your cell with ibrav=0?
Best regards
Julen
Dear Dr. Larrucea,
I have the same problem as I have defined my system with ibrav = 0 and yes there’s no alat defined. Also, putting in
grep -a CELL_PARAMETERS out |tail -1 returns
CELL_PARAMETERS (bohr)
What changes should I make in the output file to utilize this script correctly?
I have tried replacing (bohr) with (alat=lattice parameter) but it doesn’t work.
Please advise.
Dear Dr. Larrucea,
I’m sorry to have disturbed you. By replacing
CELL_PARAMETERS (bohr)
with
CELL_PARAMETERS (alat= 1.0)
the code works fine.
Thank you for the script.
Dear Julen
I have read many cool things in your website and although I didn’t understand many of them, I enjoyed having this tour! I am fresh in Quantum espresso and actually I didn’t know what is Python until I read here and then searched about it. I tried doing vc-relax calculation with the python and I got an error: (If there wasn’t any I should surprise!)
Traceback (most recent call last):
File “pw2cellvec.py”, line 28, in
fh=open(output,”r”)
IOError: [Errno 2] No such file or directory: ‘out’
That’s what I did: run a vc-relax calculation
saved your python in a file, the same path with vc-relax.out
typed : python name.py in Terminal
am I doing write?
Sorry! My english is as bad as my computer!!!! hahaha
Thanks again for your useful website.
Dear Samira,
it looks like you you have a problem with the name of the quantum espresso output file.
I used to call them always just “out” because it makes the things easier for automating tasks.
You can rename your output as “out” (so it finds the default file name) or you can use:
pw2cellvec.py -o your_output_filenameGood luck 🙂
Oh! I love you! now it’s working and it’s cool because It shows cell parameters in Angstrom!
big like and Thanks alot
Hi sir,
I was trying to relax Co2(CO)8 .but it didnot happen.the program stopped.the ecutoff was 45 and ecutrho was 180(4×45).sir please tell me the way out to relax this molecule.this was not vc relax.i am sorry i donot know python.
Hi Sapna,
I bet your geometry is too far from the relaxed structure. You might be falling into a saddle point either with the geometry or the wavefunction. Does the total energy converge somehow?
Hi
this command doesn’t work in Terminal.
I don’t have Python
Is there any other way to get lattice parameter ?
Regards